Proteases are specialized enzymes responsible for degrading damaged, misfolded, or unneeded proteins within the cell. The human mitochondrial presequence protease (hPreP) breaks down several distinct proteins, including beta amyloid (Aβ) species known to aggregate and form the amyloid plaques associated with Alzheimer’s disease. Using a combination of high-resolution x-ray crystallography and small angle x-ray scattering (SAXS) at the APS, researchers from The University of Chicago and the University of Illinois at Chicago were able to define the mechanism by which hPreP recognizes a diverse array of amyloid proteins. The findings reveal that hPreP uses a large catalytic chamber to capture “toxic” peptides on the basis of their size and charge distribution. Understanding the mechanism of hPreP substrate recognition and degradation paves the way for the development of small-molecule modulators of hPreP, which could ultimately serve as a new therapeutic approach in the treatment of Alzheimer’s disease.
Proteases are specialized enzymes responsible for degrading damaged, misfolded, or unneeded proteins within the cell. The human presequence protease (hPreP) is responsible for degrading “presequences,” which are amino acid sequences that direct newly synthesized proteins to the mitochondria. Once inside the mitochondria, however, presequences are toxic, and can interfere with the mitochondria’s ability to provide energy to the cell. In addition to degrading presequences, hPreP also degrades several Aβ proteins. Aβ peptides, when they accumulate, form the amyloid aggregates, or plaques, characteristic of Alzheimer’s disease.
Diffraction data critical for defining the crystal structure of hPreP alone and bound to Aβ were collected at the SBCCAT 19-ID-D beamline at the APS. SAXS data were collected at the BioCAT 18-ID-D beamline at the APS.
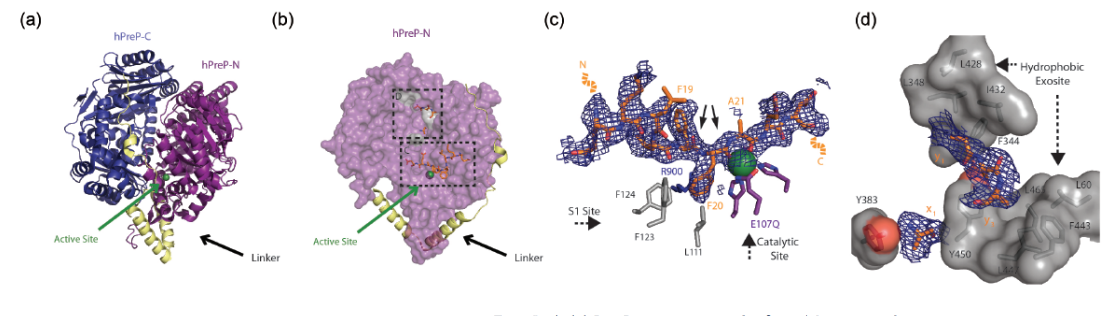
The data (Fig. 1) reveal how hPreP is able to accommodate distinct molecules within its catalytic site, and how this enzyme specifically recognizes amyloid proteins. The high-resolution diffraction patterns of hPreP alone and in complex with Aβ provide an atomic level snapshot of the large (~13,000 Å) catalytic chamber and the molecular interactions required for substrate capture and cleavage. Within this chamber, amino acid residues form a hydrophobic pocket, a basic pocket adjacent to the catalytic site, and a hydrophobic exosite distant from the catalytic site. This arrangement indicates that charge complementarity is a driving force for hPreP’s ability to bind Aβ in an orientation that permits specific cleavage of Aβ within regions likely to form aggregates. Additional SAXS data show that in solution, hPreP exists in a mixture of “open” and “closed” conformations at ~1:4 ratio. Substrate binding promotes most of the hPreP to adopt the “closed” conformation, which induces the alignment of the catalytic residues in the proper orientation. This observation confirms that hPreP excludes molecules from the catalytic site on the basis of their size.
The flexibility of hPreP substrate recognition leaves unanswered the question of whether additional, currently unidentified proteins are targeted by hPreP for degradation. Identification of hPreP substrates will illuminate additional roles hPreP has in other important cellular pathways.
See: John V. King, Wenguang G. Liang, Kathryn P. Scherpelz, Alexander B. Schilling, Stephen C. Meredith, and Wei-Jen Tang, “Molecular Basis of Substrate Recognition and Degradation by Human Presequence Protease,” Structure 22, 996 (July 8, 2014). DOI: 10.1016/j.str.2014.05.003
This research was supported by the National Institutes of Health Grant R01 GM81539 to W.J.T. BioCAT is supported by grant 9 P41 GM103622 from the National Institute of General Medical Sciences of the National Institutes of Health. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357.
Adapted from an APS press release by Emma Nichols.