Cardiomyopathies are diseases of the heart muscle in which the muscle of the pumping chamber (ventricle) can become enlarged (dilated cardiomyopathy; DCM) or thickened (hypertrophic cardiomyopathy; HCM), potentially leading to heart failure. There are currently no effective treatments but the disease often has a genetic component related to mutations in the heart muscle proteins that are involved in muscle contraction, so some researchers have focused their therapeutic development efforts on correcting these muscle contraction problems based on the structural basis of the defect. A recent study from a team of researchers using the U.S. Department of Energy’s Advanced Photon Source (APS) employed humanized mouse models expressing mutations observed in patients with HCM and DCM to evaluate the structure-function relationships and the changes observed in cardiac muscle contraction with these mutations. The work, published in the Proceedings of the National Academy of Sciences of the United States of America, provides a deeper understanding of the effects of cardiomyopathy-causing gene mutations on heart muscle contraction that could lead to the development of new therapies for this potentially life-threatening disease.
About 70% of patients with inherited HCM have a defect in the gene for the cardiac myosin protein or the cardiac myosin binding protein C. However, recent genetic evidence has suggested that mutations in the cardiac regulatory light chain (RLC) MYL2 gene are more common than previously thought and can be associated with poor outcomes. In order to understand the structural basis for how mutations in the MYL2 gene can cause HCM, or the less commonly occurring DCM, the research team performed muscle structure and force measurement experiments on heart muscle samples from mice that express mutated human RLC proteins, the HCM-D166V mutation associated with hypertrophic cardiomyopathy or the DCM-D94A mutation associated with dilated cardiomyopathy.
Normally, the cardiac regulatory light chain protein acts as a major subunit of the cardiac myosin protein to regulate calcium-mediated interactions with other muscle proteins and the myosin movements that result in the power stroke of muscle contraction. On this basis, the team set out to understand the relationship between these two mutations in the MYL2 gene and associated defects in the function of RLC in cardiomyopathy.
They used the small-angle x-ray diffraction technique at the Biophysics Collaborative Access Team 18-ID x-ray beamline at the APS to compare the spatial orientation of the thick and thin muscle filaments in left ventricular papillary muscle (LVPM) from mice expressing the mutated human RLC to mice expressing the normal, unmutated wildtype human cardiac RLC. (The APS is an Office of Science user facility at Argonne National Laboratory.)
In this model system, small-angle x-ray diffraction can measure interfilament lattice spacing that is proportional to the distance between two adjacent muscle filaments and equatorial intensity ratios that provide information about the number of myosin heads that are attached to actin-containing thin filaments (cross-bridges). By comparing structural results at different concentrations of calcium and simultaneously recording force traces, they were able to get structure-function information for all three muscle types. The intensity ratios at different calcium concentrations showed that contraction of the HCM mutant muscles were the most sensitive to calcium, forming more cross-bridges at submaximal calcium concentrations than either the DCM mutant or the wildtype muscle.
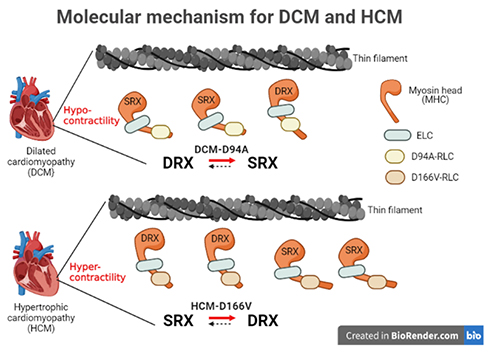
The HCM and DCM mutations also seem to affect the super-relaxed (SRX) state of myosin and the equilibrium between the SRX and the disordered relaxed state (DRX), SRX ↔ DRX, differently. The super-relaxed state is an energy-conserving state of relaxed muscle myosin that uses lower levels of ATP. Fluorescent nucleotide exchange experiments on the different mouse LVPM fibers showed that the HCM mutation destabilizes the energy-conserving SRX state and promotes the DRX state, leading to the hypercontractile behavior that was also observed in the X-ray diffraction experiments. Conversely, the DCM mutation actually favors the SRX, consistent with hypocontractile cross-bridge behavior observed with DCM (Fig. 1).
These results provide structural information about how these mutations manifest as the heart muscle problems observed in cardiomyopathy. The HCM mutation causes the protein to be more sensitive to calcium, to have higher levels of cross-bridge formation, and to be less often in the super-relaxed state, all consistent with the features of hypercontractility observed in HCM patients. The DCM mutation didn’t exhibit changes in calcium sensitivity but was more often in the super-relaxed state, consistent with the low ejection fraction dysfunction observed in DCM patients.
Understanding the structural basis for these RLC mutations may be the first step in the design of treatments that can correct the problems associated with these cardiomyopathy mutations.
See: Chen-Ching Yuan, Katarzyna Kazmierczak, Jingsheng Liang, Weikang Ma, Thomas C. Irving, and Danuta Szczesna-Cordary, “Molecular basis of force-pCa relation in MYL2 cardiomyopathy mice: Role of the super-relaxed state of myosin,” Proc. Nat. Acad. Sci. USA 119(8), e2110328119 (2022). DOI: 10.1073/pnas.2110328119